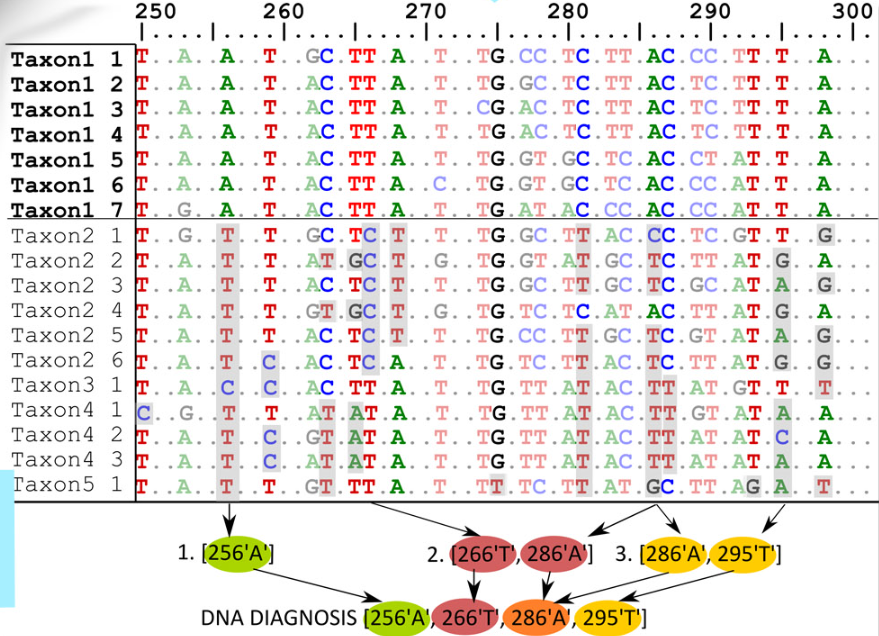
DNA sequence data are widely used in phylogenetic studies and for establishing species boundaries. At the same time, formal descriptions of new taxa are still predominantly based on traditional approaches, i.e. on morphology, and DNA features are rarely used for this purpose. However, a wider integration of DNA data into formal taxonomy can significantly improve the quality and usability of descriptions: the same features that are used to distinguish between taxa form the basis of their description. Since the acquisition of DNA sequence data is relatively inexpensive, highly productive, and standardized, DNA traits are obviously more accessible to researchers than taxonomic knowledge, especially in difficult to systematize taxa. In practice, the most significant obstacles are the lack of both a generally accepted practice of using DNA data from taxonomy, and a sufficiently powerful and flexible algorithm for identifying taxonomically important features in DNA data. Moreover, there remains a distrust of DNA features on the part of traditional taxonomists, since it is unclear whether DNA features are reliable enough to build formal descriptions of taxa on them.
A group of researchers led by a researcher at IEE RAS, Ph.D. Alexander Fedosov has developed a novel MOLD algorithm for identifying taxon diagnostic characters in monolocus (i.e., containing sequences of a single gene) DNA data. MOLD is superior to other existing algorithms for identifying diagnostic DNA features in both speed and functionality. It has been shown that currently used diagnostic DNA features are often absent, especially in datasets that include hundreds of species, or are not sufficiently reliable. An original solution has been developed - an additional algorithm that models the unexplored genetic diversity of taxa, and on its basis offers the optimal diagnostic combination of nucleotides (rDNC) in DNA data. It has been shown that the reliability of rDNC significantly exceeds the reliability of previously used DNA features. Since MOLD is the only program capable of identifying diagnostic combinations of nucleotides that meet the given reliability criteria, its use has almost no alternatives, especially when working with DNA data from a large number of taxa. MOLD is available as a Python command line application, and has a graphical interface for running in a web browser. The results of testing MOLD in comparison with previously proposed software solutions are published in the highly rated scientific journal Molecular Ecology Resources