Данные о последовательностях ДНК широко применяются в филогенетических исследованиях, а также для установления границ видов. В то же время, формальные описания новых таксонов все еще преимущественно опираются на традиционные подходы, т.е. на морфологию, и признаки ДНК используются крайне редко. Тем не менее, более широкая интеграция ДНК данных в формальную таксономию способна существенно улучшить качество и практичность описаний: те же признаки, которые используются для разграничения таксонов ложатся в основу их описания. Так как получение данных о последовательностях ДНК относительно недорого, высоко производительно и стандартизовано, признаки ДНК становятся очевидно более доступны исследователям, чем таксономическое знание, особенно в сложных для систематики таксонах. Практически наиболее значимые препятствия – отсутствие как общепринятой практики использования ДНК данных с таксономии, так и достаточно мощного и гибкого алгоритма для выявления таксономически-важных признаков в данных ДНК. Более того, сохраняется недоверие к признакам ДНК со стороны традиционных систематиков, так как неясно, достаточно ли надёжны признаки ДНК для того, чтобы строить на них формальные описания таксонов.
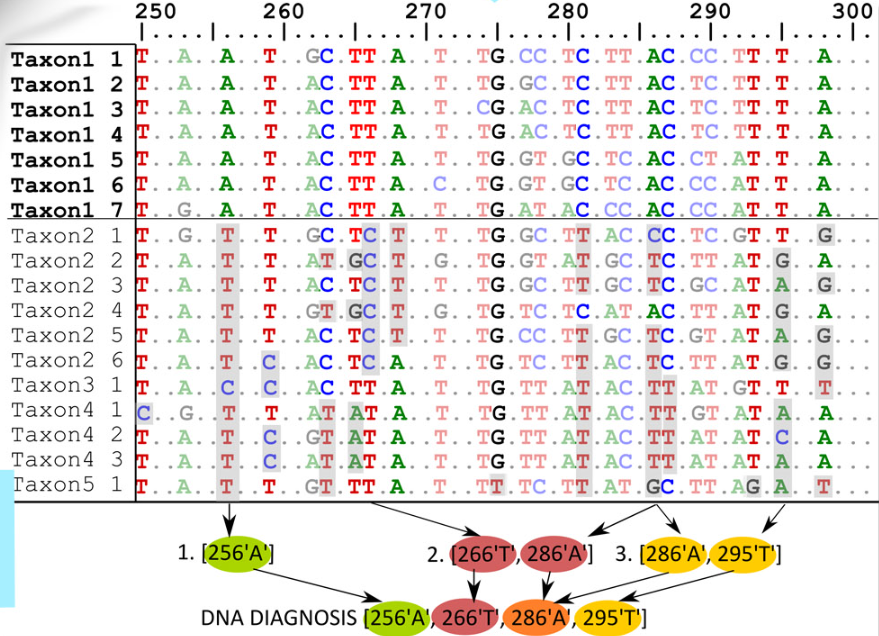
Группа исследователей во главе с научным сотрудником ИПЭЭ РАН к.б.н. Александром Федосовым разработала новый алгоритм MOLD для определения диагностических признаков таксонов в монолокусных (т.е. содержащих последовательности одного гена) данных ДНК. MOLD превосходит другие существующие алгоритмы для определения диагностических ДНК признаков как по скорости, так и по функциональности. Показано, что диагностические ДНК признаки, применяемые на данный момент, часто отсутствуют, особенно в массивах данных, включающих сотни видов, либо они недостаточно надёжны. Разработано оригинальное решение – дополнительный алгоритм, который моделирует неисследованное генетическое разнообразие таксонов, и на его основании предлагает оптимальную диагностическую комбинацию нуклеотидов (rDNC) в ДНК данных. Показано, что надёжность rDNC существенно превосходит надёжность ранее применяемых признаков ДНК. Так как MOLD – единственная программа, способная определять диагностические комбинации нуклеотидов, соответствующие заданным критериям надёжности, ее применение почти безальтернативно, особенно при работе с ДНК данными большого числа таксонов. MOLD доступен как Python приложение для работы с командной строкой, и имеет графический интерфейс для работы в интернет браузере. Результаты тестирования MOLD в сравнении с ранее предложенными программными решениями, опубликованы в высокорейтинговом научном журнале Molecular Ecology Resources (https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13590).